

May 23, 2016
How Spark and Hadoop Are Advancing Cancer Research
(Creations/Shutterstock)
The combination of Spark and Hadoop has supercharged big data analysis across many industries and use cases by lowering the barrier of entry to advanced analytics and thereby enabling data scientists to create data-driven products that weren’t previously possible. But one area where Spark and Hadoop are having an especially strong impact revolves around cancer research.
Cancer killed about 590,000 Americans last year, according to the Centers for Disease Control. That makes it the second leading causes of death in the United States, behind only heart disease, which killed 615,000. Forty-five years after President Nixon signed the National Cancer Act of 1971, which effectively was a declaration of war on cancer, progress against the collection of diseases has more or less stalled.
While an outright victory against cancer remains unlikely, there are signs that the momentum may be starting to shift in our favor. President Obama’s Precision Medicine Initiative (PMI) and Vice President Biden’s “Moonshot” program are shining the spotlight (and funneling funding) to promising approaches to fighting cancer. And a lot of the new approaches involve big data, in one way or another.
As two of the most powerful and popular products in the big data space, it’s no wonder that cancer researchers are gravitating toward Apache Spark and Apache Hadoop to tackle difficult compute-intensive tasks. Here are two examples of how the combination of Hadoop and Spark are helping to reshape the landscape in the field of cancer research.
Automatic Parallelization
One big supporter of the new parallel computing architectures like Hadoop and Spark is Jay Etchings, Arizona State University’s director of operations for research computing and a senior HPC architect. Etchings is credited with installing the first production Hadoop cluster at the university, and today the 44-node Hortonworks (NASDAQ: HDP) cluster is being used for a variety of projects, including cancer research.
Etchings says the Red Hat model of commercial open source software has been a boon for computational scientists and researchers, who previously had to hand-code a lot of their own HPC tools to get stuff to work. Etchings had first-hand knowledge of this while working in the pharmaceutical industry.
“For years I spent time tinkering with things like PLINK/SEQ and other tools and getting the appropriate libraries installed and getting stuff to work so I could actually do the science I wanted to do,” he tells Datanami. “The academic code is unreproducible, unbuildable, undocumented, unmaintained, backward incompatible [very bad] code. That’s really what it is.”
The rise of the Apache Hadoop/big data ecosystem has changed things dramatically for folks like Etchings, who are constantly looking for ways to help university researchers. Etchings appears quite happy with ASU’s Hadoop cluster, which leverages Spark, Hbase, Hive, Accumulo, and visualization tools from Tableau (NYSE: DATA).

Arizona State University’s director of operations for research computing and senior HPC architect Jay Etchings says Hadoop lets his researchers do some things much easier than using traditional HPC
“The architecture in itself is good because it gives us the robust scalability, the same sort of parallelization that we’d get in HPC, but without converting to MPI or writing in MPI. We don’t have the difficultly we had in HPC to scale horizontally,” he says. “You put something on it, and you run a job, and it’s already running across maple nodes. Even within Spark, you align a computation, you split up into RDDs, and it goes ahead and runs the mathematic computation.”
One of ASU’s projects involves analyzing the genetic diversity involved in Glioblastoma multiforme (GBM), a particularly lethal form of brain tumor. While the prognosis for those afflicted with GBM is generally poor, doctors have noticed there can be significant differences in survivability rates, and they haven’t been able to figure out why. Now researchers are beginning to discover what’s going on under the surface.
Etchings explains: “Up until about 15 years ago, we assumed a tumor cell is a tumor cell, just a nasty looking thing,” he says. “But what we found is there’s heterogeneity in tumors. They’re made up of clusters of all sorts of cells and hijacked and essentially now are growing out of control. Well, if we know the exact makeup of those cells and we also have a [genetic] sequence from the person themselves, we can tell that if both Jay and Alex come into cancer treatment center at once, maybe Jay needs surgery and intervention in a week whereas Alex can wait.”
In other project, researchers are using graph analysis techniques to discern protein pathways using naïve Bayesian analysis. Protein pathways look identical to social graphs from sites like Facebook or Google, Etchings says, so some of the same advanced analytic techniques can be used to glean insights out of them.
“Where this is going [is the use of] machine learning mechanisms where we can actually start to mine for non-obvious relationships,” he says. “If I know where 215 proteins connect to 2.5 million data points, maybe I can discern where some of the other hierarchical points attach? That would be huge. At this point what we’re doing hasn’t been engineered to that point. But I believe … we will quickly get to that point.”
The power of Spark is giving ASU researchers the capability to iterate in a manner that was impossible before. For example, in one project, ASU researchers are running run 600,000 simulations in a week across 60,000 patients. “That wasn’t passible before,” Etchings says. “The real game changer for this has been Spark.”
Outlier Detection with COPA
Another big data analytics project that’s finding success in the field of cancer research involves the Cancer Outlier Profile Analysis (COPA) algorithm, which has been successfully used to detect some of the collection of genes responsible for causing prostate cancer.
In a session at this month’s Apache Big Data conference in Vancouver, British Columbia, Mahmoud Parsian, a senior architect at Illumina, described the genesis of COPA and some of the architectural decisions that shaped its creation.
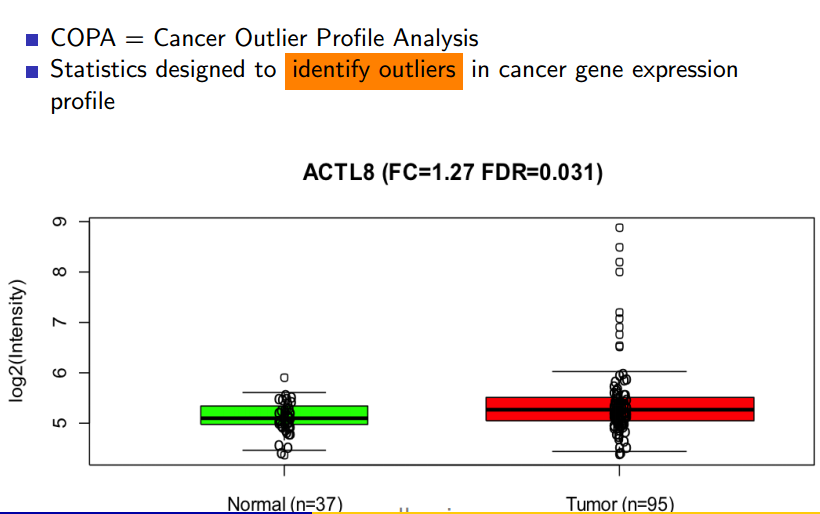
COPA has proven its value in detecting genetic mutations linked to prostate cancer, and is now being studies with other types of cancers
At its core, COPA is a technique for analyzing key-vale pairs of gene expression data and to detect outliers, which are the candidates for cancer. A parallelized algorithm was necessary owing to the size of the data involved, says Parsian, who also teaches at the University of Santa Clara.
“Of course, if the data size is small, you can detect it visually or by writing some sample programs,” Parsian says. “But when you have terabytes of data you’re analyzing, detecting mutation is impossible to do visually.”
There are many ways to solve this problem, and any number of algorithms could do the trick. Somebody may still create a better algorithm than COPA, says Parsian, who leads the big data team at Illumina. But for now, nothing has been able to beat COPA in speed and scalability.
The algorithm works by normalizing the values in a set of data, using the median absolute deviation (MAD) to segment the data, and then isolating the top 1 percent of values, which are deemed the outliers. The whole process is then run again against the outlier data set, to essentially find the outliers of the outliers. After running this result set through page-rank algorithm, the final data is output.
Running on Illumina’s 66-node Spark cluster, COPA produces good results. “We use Hadoop for storage and persistence, but for the calculations and analysis we use Spark,” Parsian says. “We used to have most of these algorithms in Hadoop MapReduce, but it was taking way too long. About two years ago we moved most of our application into the Spark cluster.”
COPA runs as a near real-time algorithm. “Our clients are wiling to wait 30 sec to 2-3 minutes to get the results for calculating this kind of operation,” Parsian says. “Maybe in a few years when servers get 2TB of RAM or so, maybe we can answer this question in a few seconds rather than a few minutes.”
Related Items:
Can Big Data Deliver on the Huge Expectations of Precision Medicine?
Big Data and the White House’s Cancer Moonshot
Technologies:
Frameworks
Vendors:
Hortonworks
Leading Solution Providers









Tabor Network
Sponsored Multimedia








Featured Events
-
AI & Big Data Expo North America 2024
 June 5 - June 6Santa Clara CA United States
June 5 - June 6Santa Clara CA United States -
CDAO Canada Public Sector 2024
 June 18 - June 19
June 18 - June 19 -
AI Hardware & Edge AI Summit Europe
 June 18 - June 19London United Kingdom
June 18 - June 19London United Kingdom -
AI Hardware & Edge AI Summit 2024
 September 10 - September 12San Jose CA United States
September 10 - September 12San Jose CA United States -
CDAO Government 2024
 September 18 - September 19Washington DC United States
September 18 - September 19Washington DC United States